
Research
-
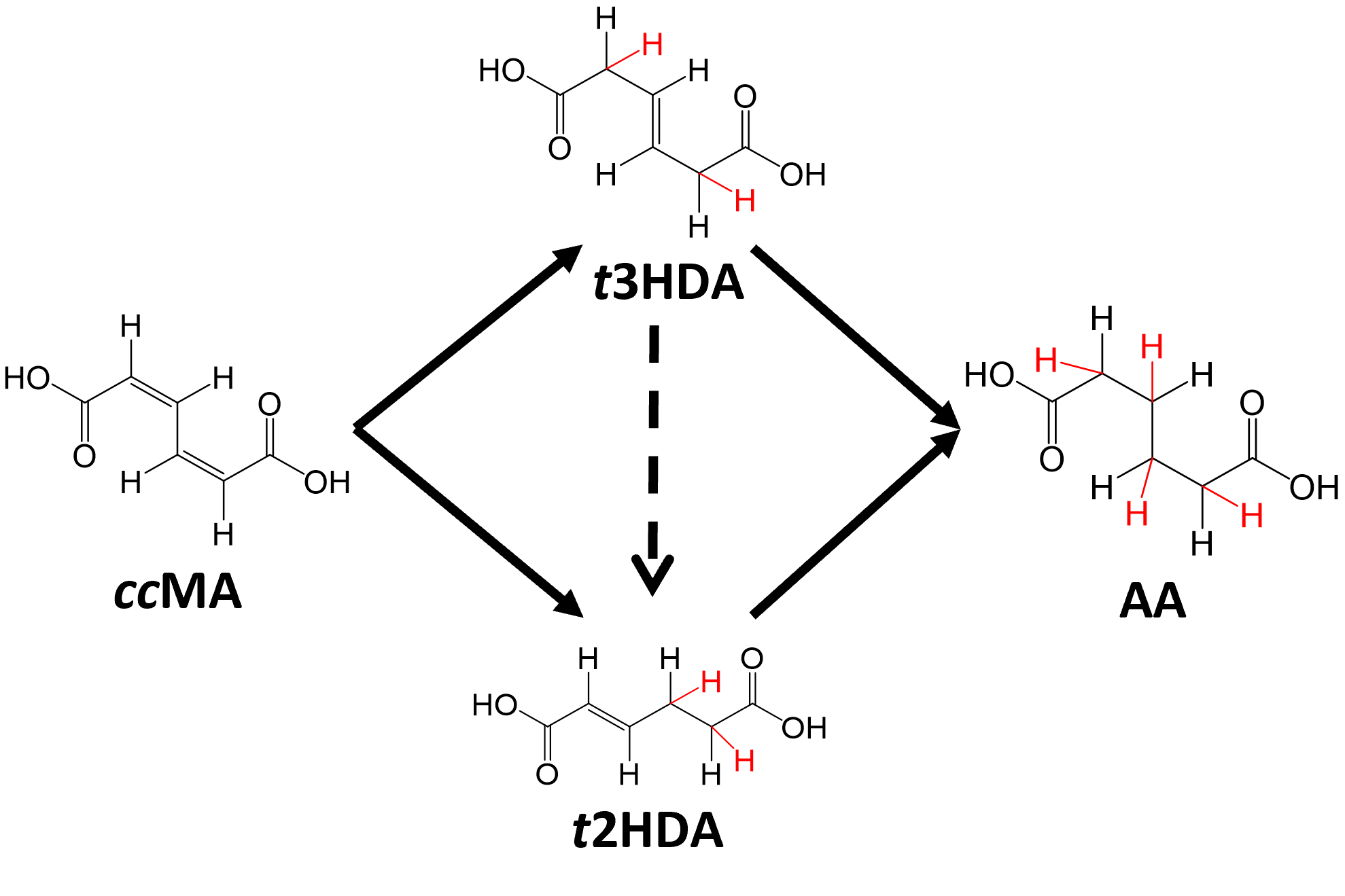
Unraveling Reaction Mechanisms
Computations are uniquely suited to uncover details of reaction mechanisms, investigating atomic-scale phenomena that cannot be directly interrogated with experimental techniques. Our group focuses on understanding the detailed mechanisms by which heterogeneous catalysts accelerate chemical transformations, uncovering atom-by-atom details of how reactions occur.
Our work focuses largely on understanding transformations of complex biomass-derived feedstocks. Such chemicals can be converted to useful platform or novel compounds through traditional (thermal) heterogeneous reactions or by using electrochemical techniques. Our atomic-scale investigations offer valuable insights into factors controlling reactivity, identifying key reaction properties for catalyst design.
-
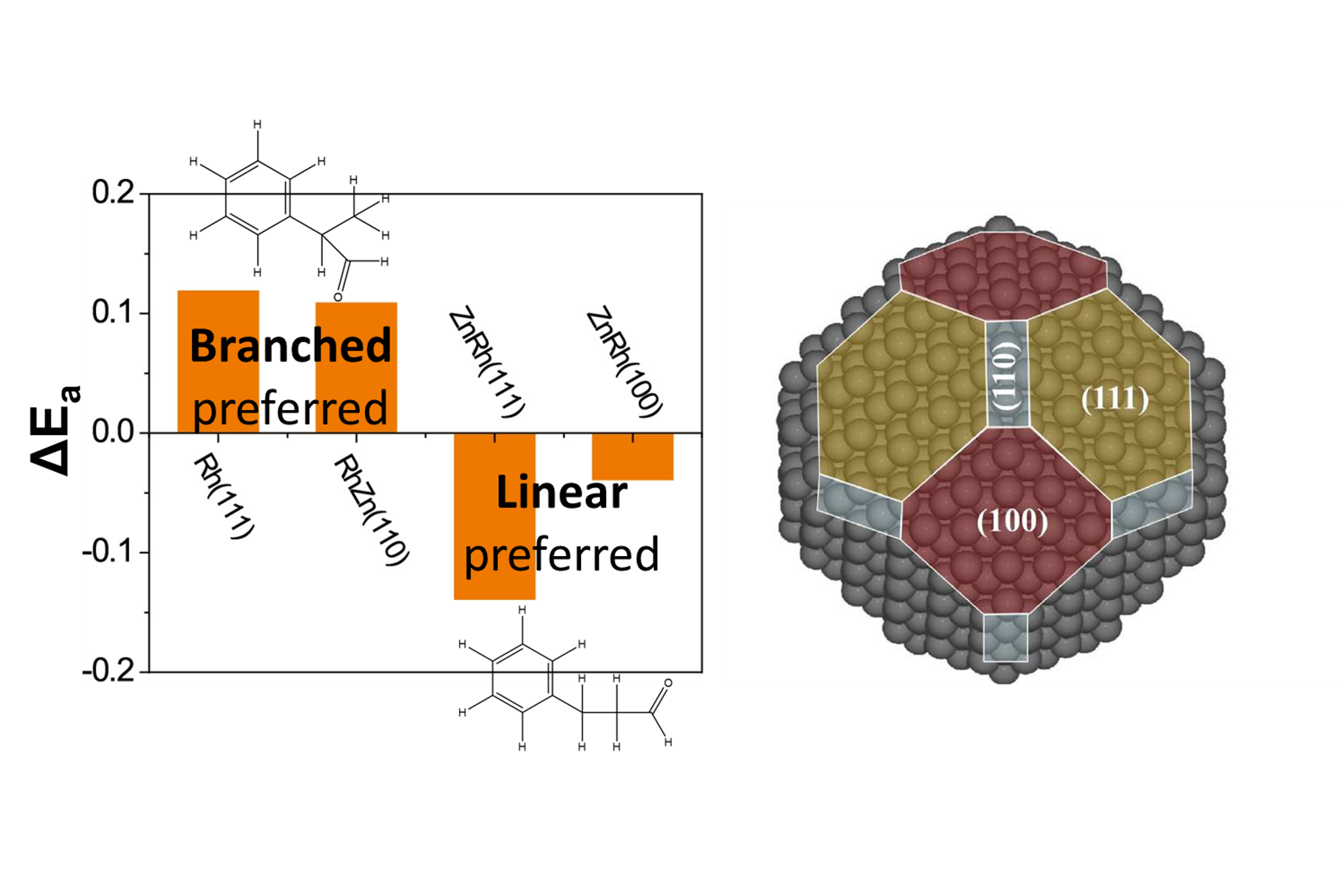
Structure-Driven Catalyst Design
The inorganic synthesis community has experienced tremendous growth in its ability to design materials. Computations are poised to take a leading role in identifying not only what elemental and alloy compositions make up an ideal catalyst, but also the key structural features that can be leveraged to maximize activity and selectivity.
Our team develops and leverages a range of structure-driven reactivity correlations to navigate the immense design space and countless atomic arrangements associated with catalyst structure. We use these insights to rapidly screen candidate structures and design catalytic active sites on an atom-by-atom basis.
-
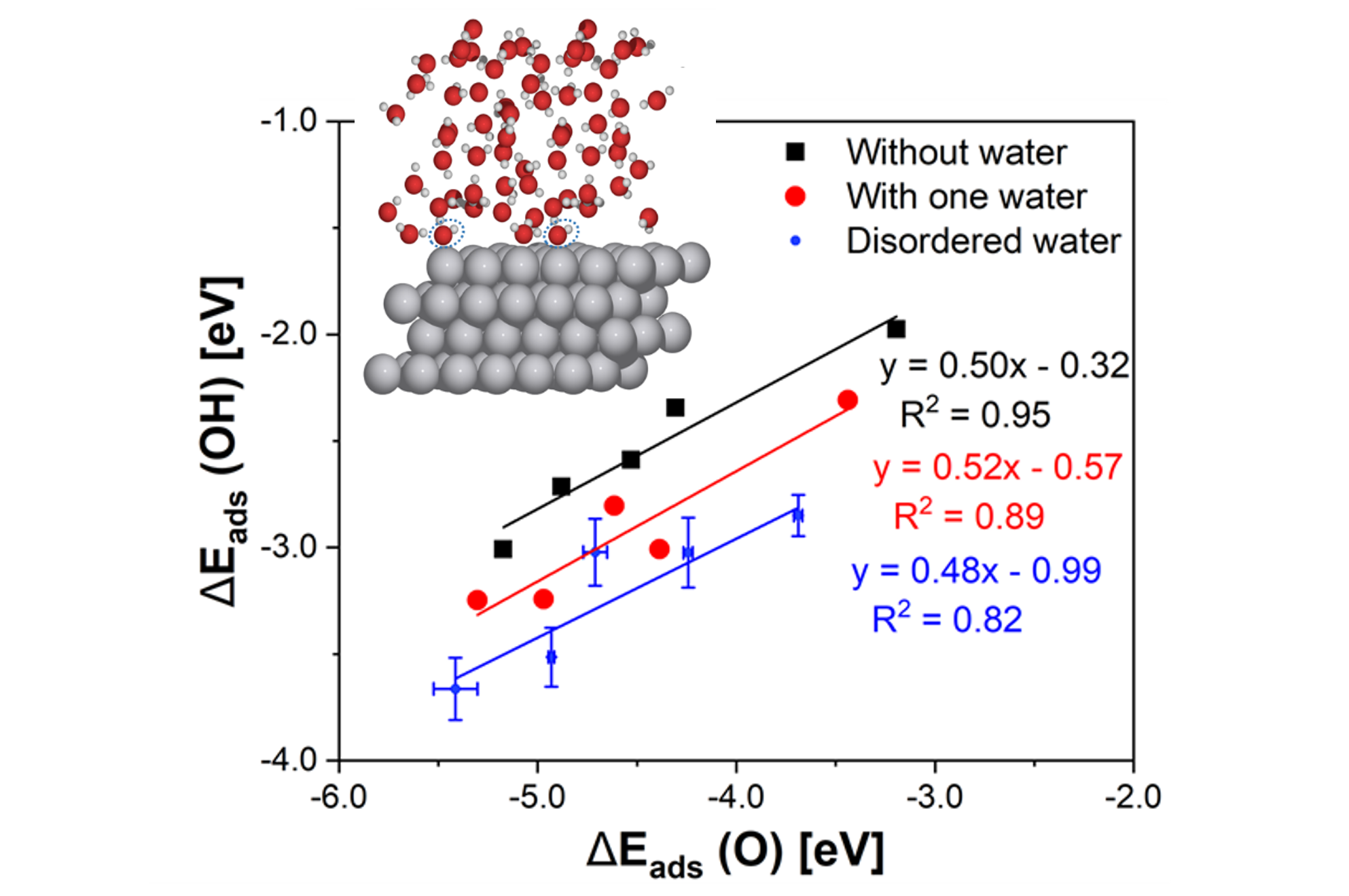
Reactivity Trends in Complex Environments
Understanding fundamental correlations between reactive species is critical to the ability to efficiently design improved materials. Computations have played a leading role in elucidating reactivity trends across a series of catalysts, enabling accelerated rational design of materials for a wide range of applications.
Our group seeks to understand the ways in which these correlations manifest in complex reactive environments, such as at the solid-liquid interfaces characteristic of many transformations of biomass-derived compounds or in electrochemistry. Our goal is to identify and exploit degrees of freedom that are not present at simpler gas-solid interfaces, providing more control in the design of active and selective catalysts.
-
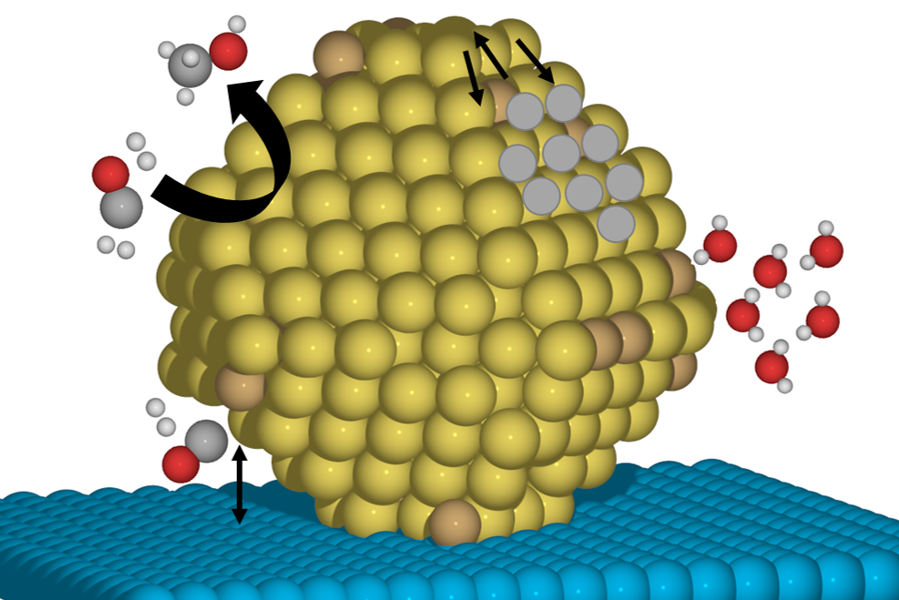
Controlling Materials Growth and Stability
To fully realize theoretically-designed features of nanostructured materials, they must be grown and maintained in what are typically thermodynamically unfavorable states. Computational simulations of materials evolution provide predictive insights into the ways in which conditions can be manipulated to maximize the lifetime and stability of materials under operating conditions.
Our research develops strategies to control the growth and evolution of both transition metal materials as well as Group IV semiconductors. By manipulating the reactive environments, we modulate the energetics associated with desirable structural features and kinetically trap materials in those desired states.